

Hoy en Revista Dosis
Mostrando artículos por etiqueta: COVID19
Miércoles, 23 Marzo 2022 13:47
Aumentaron las muertes por tuberculosis durante la pandemia
El peso real de la pandemia de COVID-19 no solo presenta como resultado el número de personas afectadas o fallecidas a causa de esta enfermedad respiratoria, sino también por el impacto que tuvo sobre otras afecciones cuya atención quedó relegada, cuando no desatendida. Es el caso de la tuberculosis –que el 24 de marzo conmemora su día mundial–: una de las enfermedades que más muertes ocasiona a nivel global y en la que se ha verificado un retroceso en cuanto a su diagnóstico y tratamiento a causa de la pandemia.
“Lo que hemos visto con la tuberculosis, al igual que en la mayoría de las patologías crónicas y prevalentes, es que su atención ha quedado relegada por la epidemia del COVID-19, porque prácticamente todos los servicios médicos nos hemos corrido de nuestras rutinas habituales para abocarnos exclusivamente a la pandemia”, advirtió el médico neumonólogo Matías Scafati, jefe de la Unidad de Internación de Neumonología del Hospital Tornú, y agregó: “Lo que estamos empezando a ver hoy son justamente las consecuencias: estas enfermedades han seguido su evolución natural”.
La tuberculosis es una enfermedad infecciosa que afecta a los pulmones y que es causada por una bacteria (Mycobacterium tuberculosis) que se transmite de una persona a otra a través de las gotitas de aerosol que permanecen en el aire tras haber sido expulsadas por personas con enfermedad pulmonar activa. Aunque en personas sanas suele ser asintomática, los síntomas de la tuberculosis pulmonar activa son: tos (a veces con esputo que puede ser sanguinolento), dolor torácico, debilidad, pérdida de peso, fiebre y sudoración nocturna.
“No todos los que estamos expuestos a la tuberculosis nos enfermamos. Una vez que ingresa a nuestro organismo, la bacteria puede ir por diferentes caminos: un camino es que nuestro sistema inmunológico la pueda eliminar por completo, el otro camino es que nuestro sistema inmunológico no lo pueda eliminar y la bacteria permanezca allí, causando lo que se denomina una infección tuberculosa latente. La bacteria queda alojada normalmente en el pulmón o en las zonas cercanas al pulmón y se puede producir una infección. Si en algún momento bajan las defensas o si se deteriora la inmunidad, se puede desarrollar esa enfermedad latente”, explicó el doctor Scafati.
Ya en octubre de 2020, a poco más de 6 meses de declarada la pandemia, la OMS publicó su informe Global Tuberculosis Report 2020, en el que advertía que el COVID-19 “amenazaba con revertir los avances recientes en la reducción de la carga global de la tuberculosis”. “El reporte más claro de tuberculosis es el que hace anualmente la Organización Mundial de la Salud –señaló el doctor Scafati–. Lo que se pone en evidencia en este reporte es que entre el 2019 y el 2020 hubo una caída significativa en la notificación de la tuberculosis de cerca del 20%: en 2019 se notificaron alrededor de 7.100.000 casos, mientras que en 2020 se notificaron menos de 6.000.000”.
En la Argentina, las estadísticas más recientes sobre la tuberculosis son previas a la pandemia. En 2019 se notificaron 12.499 nuevos casos, dando lugar a una tasa de notificación de 27,8 por cada 100.000 habitantes. Es de esperar que en los siguientes boletines los números de notificaciones sean menores, pero no por una reducción de los contagios sino porque muchos pacientes no accedieron al diagnóstico a causa la situación del sistema de salud durante la pandemia de COVID-19.
Pero a nivel global hay datos aún más preocupantes que sugieren una caída en el acceso y la adherencia al tratamiento de la tuberculosis: “Contrastando con una menor notificación de casos, aparece un aumento en la incidencia de la mortalidad, la cual venía decayendo” dijo el especialista. Según las estadísticas más recientes de la OMS, mueren hoy 1.5 millones de personas al año a causa de la tuberculosis, lo que la convierte en la enfermedad infecciosa que más muertes ocasiona en el mundo.
El Día Mundial de la Tuberculosis fue establecido en conmemoración del día en el que Robert Koch anunció que había descubierto la bacteria que causa la tuberculosis en 1882, lo que abrió el camino hacia el diagnóstico y la cura de esta enfermedad.
Publicado en
Top
Lunes, 07 Marzo 2022 10:51
La OMS aprobó el molnupiravir, primer tratamiento oral contra el Covid-19

La Organización Mundial de la Salud (OMS) anunció la inclusión del antiviral molnupiravir en su lista de tratamientos recomendados para enfermos de covid-19. Se trata del primer fármaco de administración oral recomendado por la entidad contra el coronavirus.
La OMS recomienda su utilización solo entre pacientes de covid-19 que no hayan desarrollado formas graves de la enfermedad, pero que corran un muy alto riesgo de hospitalización, tales como personas no vacunadas, de la tercera edad, pacientes con deficiencias inmunológicas o con enfermedades crónicas. Al mismo tiempo, el organismo internacional desaconseja su uso entre niños y mujeres embarazadas o lactantes.
El fármaco, desarrollado por el laboratorio estadounidense MSD (Merck Sharp & Dohme), se consume en pastillas y, según la OMS, si se utiliza con los primeros síntomas de infección, puede evitar hospitalizaciones. Esta medicación se administra en cuatro pastillas (800 miligramos) dos veces al día durante cinco jornadas, el tiempo habitual de desarrollo de síntomas de la enfermedad. Usado lo antes posible después de la infección, indica la OMS, puede ayudar a prevenir la hospitalización.
El medicamento actúa introduciendo errores en el código genético del virus SARS-CoV-2, lo que evita que el virus se replique en el organismo. La recomendación se basa en nuevos datos de seis ensayos en los que participaron 4.796 pacientes. Este es el conjunto de datos más grande sobre este fármaco que existe hasta el momento. También se informó que si bien molnupiravir no está ampliamente disponible, se tomaron medidas para mejorar las posibilidades de acceso a este remedio. Todavía no está disponible en Argentina.
Médicos Sin Fronteras alertó ya en 2021 de que un tratamiento de cinco días con molnupiravir a precios de mercado puede costar unos 700 dólares, aunque si se desarrollaran genéricos alternativos, como ya se hace en India, su precio podría bajar a 20 dólares. Por otra parte, la novena actualización de la guía terapéutica para el covid-19 de la OMS incluyó una recomendación sobre casirivimab-imdevimab, un cóctel de anticuerpos monoclonales.
En base a la evidencia de que esta combinación de medicamentos es ineficaz contra la variante Ómicron del covid-19, la OMS ahora recomienda que solo se la administre ante infecciones causada por otras variantes.
Países que lo aprobaron
El antiviral molnupiravir está aprobado en Reino Unido, Estados Unidos, Japón e India. Y también en varios países de Latinoamérica. México le otorgó una autorización de emergencia en enero. Ese mismo mes, Bolivia comenzó a comercializarlo, mientras que Perú autorizó el pasado 17 de febrero su comercialización. El gobierno de El Salvador anunció, también en febrero, la compra de 4 millones de dosis del fármaco, que ya comenzó a distribuir. Este miércoles, en tanto, el Ministerio de Salud Pública (MSP) de Uruguay confirmó la habilitación del molnupiravir a la vez que aclaró que, de todas formas, el medicamento aún no está regulado en el país.
El mes pasado, el laboratorio indio Hetero informó que logró demostrar que la droga molnupiravir evita en un 65% las hospitalizaciones en pacientes con coronavirus moderado a grave de acuerdo a los estudios en fase tres presentados en la Conferencia de Retrovirus e Infecciones Oportunistas.
Publicado en
Noticias
Viernes, 04 Marzo 2022 12:43
Vitamina D2 ó Vitamina D3, cuál es la mejor?

Una nueva investigación halló diferencias significativas entre los dos tipos de vitamina D, y concluyó que la vitamina D2 tiene un impacto "cuestionable" en la salud humana. Por el contrario, encontró que la vitamina D3 podría ayudar a equilibrar el sistema inmunológico y a fortalecer las defensas contra infecciones virales como el COVID-19..
En un estudio colaborativo de las universidades británicas de Surrey y Brighton, los investigadores investigaron el impacto de los suplementos de vitamina D (D2 y D3) tomados diariamente durante un período de tres meses. Los resultados fueron publicados en la revista Frontiers in Immunology.
El equipo de investigación descubrió que ambos tipos de vitamina D no producían el mismo impacto. Encontraron evidencia de que la vitamina D3 tenía un efecto modificador en el sistema inmunológico que podría ayudar al organismo a defenderse contra enfermedades virales y bacterianas.
"Hemos demostrado que la vitamina D3 parece estimular el sistema de señalización del interferón tipo I en el cuerpo, una parte clave del sistema inmunitario que proporciona una primera línea de defensa contra las bacterias y los virus. Por lo tanto, un estado saludable de vitamina D3 puede ayudar a prevenir que virus y bacterias se afiancen en el cuerpo", explicó sobre los hallazgos el profesor Colin Smith, autor principal del estudio.
"Nuestro estudio sugiere que es importante que las personas tomen un suplemento de vitamina D3 o alimentos adecuadamente fortificados , especialmente en los meses de invierno", subrayó Smith.
La vitamina D3 se produce naturalmente en la piel a partir de la exposición a la luz solar o a la luz ultravioleta UVB artificial, mientras que algunas plantas y hongos producen vitamina D2.
Muchas personas tienen niveles insuficientes de vitamina D3 porque viven en lugares donde la luz solar es limitada en invierno, como el Reino Unido. Al pasar más tiempo en casa, la pandemia de Covid-19 también limitó la exposición natural de las personas al sol.
"La deficiencia de vitamina D puede deberse al área geográfica donde vivimos (el sur de nuestro país tiene menores niveles de vitamina D), al factor estacional (ya que el invierno, el frío y la lluvia disminuyen la radiación solar), a la contaminación ambiental o al hábito de estar muchas horas sin salir a la luz solar", explicaba en una nota con Clarín Alicia Bagur, médica osteóloga, directora médica de Mautalen Salud e Investigación.
Aunque algunos alimentos están fortificados con vitamina D (cereales para el desayuno, yogures y pan) y pocos c, pocos contienen la vitamina de forma natural (pescados grasos como el salmón, boquerón, caballa o arenque; la yema de huevo; los hongos expuestos al sol por 30 a 60 minutos; y lácteos suplementados con vitamina D).
El colecalciferol (vitamina D3) pertenece a una clase de medicamentos llamados análogos de la vitamina D. El cuerpo necesita el colecalciferol para tener huesos, músculos y nervios saludables y fortalecer el sistema inmunitario. Su acción consiste en ayudar al cuerpo a usar más del calcio que se encuentra en los alimentos o suplementos.
"Si bien descubrimos que la vitamina D2 y la vitamina D3 no tienen el mismo efecto sobre la actividad genética en los humanos, la falta de impacto que encontramos al observar la vitamina D2 significa que se requiere en forma urgente un estudio más amplio para aclarar las diferencias en los efectos. Sin embargo, estos resultados muestran que la vitamina D3 debería ser la forma preferida para los alimentos y suplementos fortificados", concluyó la profesora Susan Lanham-New, coautora del estudio y jefa del Departamento de Ciencias de la Nutrición de la Universidad de Surrey.
Publicado en
Top
Martes, 15 Febrero 2022 11:02
¿Quiénes recibirán la cuarta dosis en Argentina?
A nivel mundial, el 55% de la población recibió el esquema completo de vacunación contra el COVID-19. En la Argentina, más del 78% de la población general ya accedió a ese esquema primario al aplicarse las dos dosis de vacunas. Además el 33,1% recibió una tercera dosis como refuerzo o como adicional. En marzo próximo, se empezará a aplicar una cuarta dosis solo en las personas que han recibido la dosis adicional, según el plan estratégico de vacunación del Ministerio de Salud de la Nación.
Esa cuarta dosis ya se había establecido para un grupo específico de la población en el país. En noviembre pasado, la cartera de Salud había especificado en los lineamientos de vacunación que se iba a dar una dosis adicional como parte del esquema primario, cuando la respuesta inmune inicial fuera “probablemente insuficiente”.
Dentro del grupo de los que han recibido la tercera dosis como adicional, están las personas con inmunocompromiso. Se incluyó a las personas en tratamiento oncológico para tumores sólidos y onco-hematológicos, las que han recibido un trasplante de órgano sólido en tratamiento inmunosupresor, y las que recibieron trasplante de células progenitoras hematopoyéticas en los últimos 2 años o en tratamiento inmunosupresor.
También dentro del grupo de los que recibieron la dosis adicional están las personas con inmunodeficiencia primaria moderada o grave, personas que viven con VIH independientemente del recuento de CD4 y niveles de carga viral, personas en tratamiento activo con corticosteroides en dosis altas o medicación inmunosupresora, con insuficiencia renal crónica en hemodiálisis, con enfermedades autoinmunes y/o tratamientos con inmunosupresores, inmunomoduladores o biológicos.
Se incluyó también en el grupo de dosis adicional a las personas de 50 años o mayores que habían recibido un esquema primario de vacuna a virus inactivado, como la desarrollada por Sinopharm.
Después de los cuatro meses de haber recibido la dosis adicional, ese grupo de personas accederá entonces a la cuarta dosis que cumple la función de dosis de refuerzo como se viene dando en el resto de la población.
“Las únicas personas que recibirían una cuarta dosis son aquellos que por indicación recibieron dosis adicional en noviembre o en diciembre. Es decir, lo que reciben es el refuerzo pasado cuatro meses de la dosis adicional”, dijo Juan Manuel Castelli, Subsecretario de Estrategias Sanitarias del Ministerio de Salud de la Nación y a cargo del plan de vacunación. En marzo, se cumplirán los 4 meses de intervalo para las personas que recibieron la dosis adicional en noviembre.
La "dosis de refuerzo" para Covid-19 se aplica por una razón diferente a la "dosis adicional". Según la cartera de Salud, el refuerzo (o “booster”) se administra luego del esquema primario con respuesta inmunológica inicial suficiente. Tiene en cuenta la posible disminución de la respuesta en el transcurso del tiempo.
En el país, otras vacunas también implican la aplicación de dosis de refuerzo como por ejemplo la triple viral y la doble bacteriana.
Las tercera dosis -tanto las que se aplican como adicionales como las de refuerzo- se habían empezado a dar en noviembre pasado.
De a poco, las terceras dosis pasaron a ser la mayoría de las dosis semanales del plan de vacunación. En la primera semana de enero pasado, el 64% del total aplicado se usó como adicional o como refuerzo. En la última semana de enero, la proporción de aplicaciones de la tercera dosis (incluyendo las dosis como adicionales y las de refuerzo) pasó a ser el 69% .
Publicado en
Noticias
Martes, 01 Febrero 2022 13:02
Se confirmó el primer caso de Ómicron Sub-linaje BA.2 en Argentina

En el día de ayer se confirmó por parte del Instituto Nacional de Enfermedades Infecciosas ANLIS Malbrán el primer caso de variante Ómicron sub-linaje BA.2 en Argentina.
Se trata de un paciente de 62 años, que reside en la ciudad de Buenos Aires, que volvió el 12 de enero de Uruguay y dos días después comenzó a presentar síntomas compatibles con la enfermedad. El día 19 de enero se le realizó un test diagnóstico para SARS-CoV-2 que arrojó un resultado positivo. En todo momento, el paciente cursó la enfermedad de manera leve y fue seguido de manera ambulatoria. Entre sus contactos estrechos se encuentra su esposa, que también presenta síntomas compatibles con COVID-19.
Situación internacional:
El primer aislamiento de la nueva variante Ómicron fue reportado el 11 de noviembre en Botswana. Apenas dos semanas después, el 26 de noviembre de 2021, la OMS designó la variante B.1.1.529 como una variante de preocupación. Esta variante incluye 4 sub linajes Pangolin: B.1.1.529, BA.1, BA.2 y BA.3.
En ese sentido, análisis preliminares han evaluado que el sub linaje BA.2, recientemente detectado en el país, tendría una mayor tasa de crecimiento en comparación con BA.1. Además, sugieren que la tasa de ataque secundaria entre contactos convivientes sería mayor que entre los contactos de otros sub-linajes de Ómicron. Por otro lado, BA.2 podría asociarse con una mayor susceptibilidad a la infección.
Sin embargo, datos preliminares provenientes de estudios realizados en Reino Unido no hallaron diferencias entre la efectividad frente a enfermedad sintomática de la vacunación con esquema completo más dosis de refuerzo entre los sub linajes BA.1 y BA.2. Todos estos hallazgos requieren aún mayores investigaciones.
En la actualidad, la epidemiología genómica global se caracteriza por un predominio de la variante Ómicron, en paralelo a una disminución continua en la prevalencia de la variante Delta y una muy baja circulación de las variantes Alpha, Beta y Gamma.
Al día 25 de enero de 2022, entre las 372.680 secuencias registradas a GISAID con muestras recolectadas en los últimos 30 días, 332.155 (89,1%) pertenecen a Ómicron, en tanto, 39.804 (10,7%) corresponden a Delta, 28 (<0,1%) Gamma, cuatro (<0,1%) Alpha y dos (<0,1%) otras variantes circulantes (VOIs Mu y Lambda).
Al mismo tiempo, el sublinaje BA.1 representó el 98,8% de las secuencias registradas en GISAID, aunque varios países han informado recientemente un aumento en la proporción de secuencias BA.2, entre ellos Dinamarca, India, y el Reino Unido.
Recomendaciones para la población:
-Iniciar o completar los esquemas de vacunación de acuerdo a las recomendaciones vigentes:
-Todas las personas a partir de los 3 años deben tener dos dosis de vacuna contra la COVID-19.
-Personas de 50 años y más que hayan recibido como esquema primario vacuna Sinopharm y aquellas personas 3 años y mayores inmunocomprometidos deben recibir una dosis adicional para completar su esquema.
-Aquellas personas que hayan recibido la segunda dosis de la vacuna hace por lo menos 4 meses deberán recibir dosis de refuerzo, de acuerdo a los planes de vacunación provinciales.
-Usar barbijo en lugares cerrados y al aire libre cuando se está cerca de otras personas no convivientes.
-Mantener una ventilación cruzada y continua de los ambientes compartidos con personas no convivientes (reuniones sociales, trabajo, escuela, espacios recreativos y todo otro espacio cerrado compartido.
-Lavarse frecuentemente las manos con agua y jabón.
-Consultar de manera temprana ante la presencia de uno o más síntomas compatibles con COVID-19.
Publicado en
Noticias
Lunes, 31 Enero 2022 12:30
Llegan los autotest a las farmacias para venta al público

Las pruebas de autotest de coronavirus llegarán este lunes a las farmacias de la ciudad de Buenos Aires y el Gran Buenos Aires, donde ya podrán venderse al público, en tanto que durante el fin de semana se realizó la distribución en localidades del interior del país, como Mar del Plata, Tandil o Rosario, informó la presidenta de la Confederación Farmacéutica Argentina (Cofa), Isabel Reinoso.
"El viernes comenzó la distribución a las droguerías y el sábado comenzó a llegar a las primeras farmacias del interior (Mar del Plata, Tandil, Bahía Blanca, Rosario) y hoy se completa la distribución a las droguerías que abastecen al Gran Buenos Aires y CABA", indicó Reinoso en una entrevista.
Y continuó: "Apenas lleguen a la farmacia van a estar disponibles ya que las farmacias ya tienen el sistema informático instalado para poder hacer la dispensa de los test de autoevaluación".
La titular de la Cofa describió que una vez realizada la dispensa, el farmacéutico toma los datos (nombre y apellido, DNI, localidad y teléfono de contacto), y en la computadora de la farmacia se imprime esa información y un código QR que el cliente puede leer desde la cámara de su celular.
"Allí se le van a desplegar cuatro botones: positivo, negativo, inválido o lo haré más adelante. La persona elige la opción que corresponde y esa información le llega directamente a la farmacia", dijo Reinoso y señaló que "si no quiere hacerlo de esa manera, estarán disponibles los métodos convencionales como avisar por teléfono, por mail, personalmente".
Asimismo, recordó que "se trata de test de antígenos rápidos y lo que determinan es si hay presencia de las proteínas del virus; si da positivo, el margen de error es bajísimo".
En referencia a cuándo utilizarse, indicó que "si una persona se entera de que es contacto estrecho se recomienda que espere un lapso de tres o cuatro días desde que vio al caso confirmado para esperar que la carga viral suba y el virus pueda ser detectado".
El autotest está indicado para pacientes mayores de 60 años, con comorbilidades, embarazadas, etc.
En relación al procedimiento, la titular de Cofa indicó que "lo primero es lavarse las manos y colocar el cassette en un lugar limpio; luego hay que pasar el hisopo cuatro o cinco veces en la narina (no más de 2 centímetro), no hace falta llegar hasta el fondo de la nariz porque son test nasales, no nasofaríngeos".
Además, indicó que el test que se comenzó a distribuir hoy puede hacerse también por saliva: "Para esto es importante tener en cuenta algunos aspectos como no haberse cepillado los dientes ni ingerido comida ni bebida, por lo que lo ideal es hacerlo a la mañana", finalizó.
Durante enero, la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (Anmat) aprobó el uso de seis test de autoevaluación.
Una vez obtenida la respuesta del comprador del test, la farmacia reportará el resultado en el Sistema Nacional de Vigilancia de la Salud.
Publicado en
Noticias
Lunes, 31 Enero 2022 09:48
Efluvio telógeno: la nueva consecuencia del Covid-19

El hallazgo se desprende de un informe de la Academia Estadounidense de Dermatología, que refleja la pérdida de cabello como un nuevo síntoma que atraviesan quienes están contagiados de coronavirus cuando transitan la última etapa previa al alta.
Aníbal Parigini, médico del Servicio de Dermatología del Hospital Universitario Austral, explicó cuáles son los síntomas, las causas y el tratamiento para esta afección. Según lo definió, “el efluvio telógeno es un tipo de caída de pelo que se produce por una alteración en el ciclo normal del crecimiento”.
Esto se produce cuando por determinado desencadenante, una cantidad anormal de folículos pasan a la etapa de reposo -una de las tres por las que transita cada pelo-. Cuando esto sucede, se genera una caída que puede incluir entre 30 y 100 pelos por día según cada persona.
Las causas posibles son múltiples e incluyen deficiencias vitamínicas y nutricionales, trastornos hormonales, enfermedades sistémicas e infecciones en la piel, entre otras. Sin embargo, la relación con el Covid-19 tiene que ver con una infección aguda causada por un episodio de fiebre fuerte.
Además, esta afección también puede ser generada por un “evento emocionalmente estresante”. En el caso de contagiarse, el estrés suele llegar encadenado el aislamiento y todo lo que implica transitar la enfermedad.
Usualmente, “la caída comienza unos 2 a 3 meses luego del evento desencadenante” y suele tratarse de casos transitorios que finalizan cuando se da la repoblación del cuero cabelludo.
A pesar de la norma, en algunas ocasiones la caída “puede hacerse crónica o recurrente”. En el caso de que requiera tratamiento, el dermatólogo aclaró que se busca eliminar el desencadenante que causó el problema.
Por este motivo, es importante controlar el estrés en caso de haber sufrido efluvio telógeno. De cualquier manera, ya sea por Covid-19 o por alguno de los motivos antes mencionados, Parigini remarcó la necesidad de consultar con un dermatólogo “para encausar la conducta por seguir y calmar la ansiedad o angustia que este cuadro pudiera generar”.
Publicado en
Noticias
Viernes, 28 Enero 2022 13:30
La píldora de Pfizer contra el Covid-19 fue aprobada en Europa

La Agencia Europea de Medicamentos (EMA) aprobó la píldora anticovid del laboratorio Pfizer, que se convierte en el primer tratamiento oral contra el covid-19 autorizado en la Unión Europea (UE).
“El Comité de Medicamentos de Uso Humano (CHMP) de la EMA ha recomendado la concesión de una autorización condicional de comercialización para el medicamento antiviral oral Paxlovid”, declaró el regulador europeo en un comunicado.
La EMA “recomendó autorizar el Paxlovid para el tratamiento del covid-19 en los adultos que no necesitan asistencia respiratoria pero que presentan un riesgo de que la enfermedad se agrave”.
Los antivirales actúan reduciendo la capacidad de un virus para replicarse, frenando así la enfermedad.
Son muy esperados porque son fáciles de administrar, pudiendo ser simplemente ingeridos con un vaso de agua.
Pfizer había declarado en diciembre que su píldora anticovid reducía en casi 90% las hospitalizaciones y muertes en personas de riesgo cuando era ingerida en los primeros días después de la aparición de los síntomas.
La EMA evaluó los datos de un estudio en el que participaron pacientes con covid-19 que demostraron que “el tratamiento con Paxlovid redujo significativamente las hospitalizaciones o muertes en pacientes que presentaban al menos una enfermedad subyacente que los exponía a un riesgo de contraer un covid-19 grave”.
La mayoría de los pacientes del estudio estaban infectados con la variante Delta, indicó la Agencia Europea de Medicamentos, pero señaló que según las pruebas de laboratorio, Paxlovid debería seguir siendo eficaz contra Ómicron.
La píldora Pfizer es una combinación de una nueva molécula, PF-07321332, y ritonavir, un antiviral contra el VIH, que se toman en forma de comprimidos separados.
“El Comité de Medicamentos de Uso Humano de la EMA concluyó que los beneficios del medicamento son mayores que sus riesgos para el uso aprobado y enviará sus recomendaciones a la Comisión Europea (CE) para una decisión rápida, aplicable en todos los Estados miembros de la UE”.
Este procedimiento de aprobación por parte de la CE suele llevar algunas horas o días.
“Con la autorización de Paxlovid esta semana, se han autorizado 6 medicamentos anticoronavirus en el marco de la estrategia terapéutica de la UE”, dijo en otro comunicado la comisaria europea de Salud y Seguridad Alimentaria, Stella Kyriakides.
Estados Unidos, Canadá e Israel forman parte de un puñado de países que ya han dado luz verde al tratamiento de Pfizer.
Publicado en
Noticias
Jueves, 27 Enero 2022 11:11
Nueva ola de Covid-19: la importancia de la desinfección

Tras la explosión de la nueva ola por casos de Coronavirus en el país, la pandemia se vuelve a instalar fuerte y los expertos enfatizan sobre las medidas que se deben tomar para la prevención y el cuidado de la salud. Si bien, el uso del barbijo y el lavado de manos siguen siendo algunas de las recomendaciones más populares, las personas se preguntan si deben volver a desinfectar absolutamente todo como al inicio.
Aunque se sabe que gran parte de las infecciones de COVID-19 ocurren debido a la transferencia aérea, Funcei recomienda que aquellos objetos que se utilizan habitualmente sean desinfectados con los productos correspondientes, como por ejemplo, el teclado y mouse de la computadora, el dispensador de alcohol y el gel, las sillas, el asiento del toilette, la tapa del inodoro, el celular, la superficie de los escritorios y los picaportes.
Con el fin de continuar contribuyendo a detener la propagación de COVID-19 dentro de los lugares cerrados, Ayudín la marca de cuidado del hogar más elegida por los argentinos y Funcei, brindan algunas recomendaciones:
· Al presentar algunos de los síntomas compatibles con Covid-19, realizar la consulta médica y mantener el aislamiento según las recomendaciones vigentes.
· Mantener distanciamiento social de al menos 2 metros.
· En el hogar, ventilar los ambientes constantemente y de manera cruzada, por ejemplo, al mantener abiertas una ventana y una puerta que estén enfrentadas.
· Usar barbijo o tapaboca mientras estás en espacios cerrados con aglomeración de personas y sin consumir alimentos o bebidas.
· No compartir vasos, cubiertos, platos ni mates, e identificalos para evitar confusiones.
· Siempre contar en la mesa con alcohol en gel, una solución de alcohol al 70% o toallitas desinfectantes.
· Limpiar y luego desinfectar los espacios antes y después de la reunión. Para ello, utilizar productos que no sólo sean efectivos contra la suciedad, sino que también contengan un activo que asegure eliminar el 99,9% de los gérmenes, como por ejemplo la lavandina o toallitas desinfectantes.
· Evitar apoyar bolsos, carteras o mochilas en las mesas del comedor o la cocina; podrían transportar microbios y, de esta manera, propagarlos.
· Higienizar las manos con agua y jabón durante 20 segundos o, si no están visiblemente sucias, con soluciones a base de alcohol. Prestar especial atención a ambas caras de las manos, al espacio entre los dedos y debajo de las uñas; sacarse los anillos antes de higienizarlas.
· Al toser o estornudar, cubrirse la boca y la nariz con el codo flexionado o con un pañuelo descartable. Luego, desechar el pañuelo inmediatamente y lavarse las manos.
· Evitar tocar los ojos, la nariz y la boca.
· Recordar vacunarte y aplicarte las dosis correspondientes.
La pandemia no terminó. Por eso, es importante actuar con conciencia y responsabilidad y seguir con los cuidados entre todos para evitar contagios. Sea verano o invierno, en contexto de pandemia, cualquier viaje y actividad grupal acarrea el riesgo de contraer o de transmitir COVID-19.
--
Publicado en
Noticias
Martes, 25 Enero 2022 09:47
Serocovid-Federal: nuevo kit de producción local para detectar Covid-19
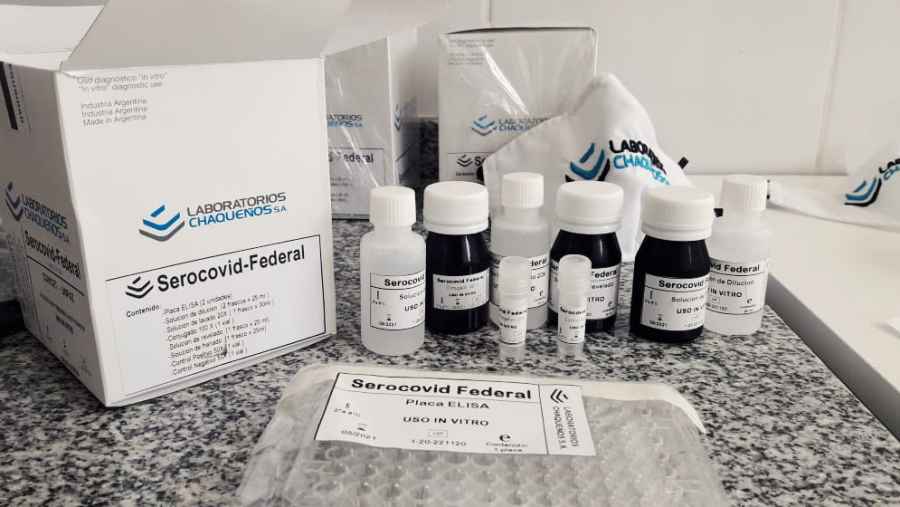
La Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT) registró el kit Serocovid-Federal desarrollado por dos expertas del Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) que detecta anticuerpos totales contra el SARS-CoV-2 y posee un 97 por ciento de eficacia. La producción se realizará en el laboratorio público Chaqueños S.A.
Se trata de un kit que utiliza la técnica Elisa que se llevó a cabo a través de un trabajo articulado entre la Universidad Nacional de José C. Paz (UNPAZ) donde se dio lugar a la investigación y al desarrollo; el Instituto Nacional de Tecnología Agropecuaria (INTA) donde se continuó su desarrollo; el laboratorio público Chaqueños S.A, en el que realizó el escalado y la producción del mismo; la Facultad de Ciencias Exactas y Naturales de la Universidad de Buenos Aires (FCEN-UBA) proveedora de la proteína recombinante que contiene secuencias del dominio RBD de SPIKE de SARS-CoV-2 ; la Administración Nacional de Laboratorios e Institutos de Salud “Dr Carlos G. Malbrán” en donde se realizó el testeo, y el mencionado laboratorio Chaqueños S.A.
Tanto el Ministerio de Salud, a través de la Agencia Nacional de Laboratorios Públicos (ANLAP), como el Ministerio de Ciencia, Tecnología e Innovación, a través de la Agencia Nacional de Promoción de la Investigación, el Desarrollo Tecnológico y la Innovación (Agencia I+D+i), fueron trascendentales en el acompañamiento, promoción y vinculación estratégica entre los distintos actores, y en el financiamiento del proyecto.
“En el marco de la pandemia de COVID-19, el Estado argentino puso en marcha acciones destinadas a promover y apoyar una asociación estratégica entre el sector académico y el sector de producción pública de medicamentos a fin de disponer de un insumo clave para el combate del COVID-19. Es un avance en materia sanitaria y un paso adelante en la reconstrucción de la matriz productiva de nuestro país. Un proyecto público con mirada federal y perspectiva de género”, explicó la presidenta de la Agencia Nacional de Laboratorios Públicos, Ana Allemand.
Publicado en
Noticias


Lo más visto
- Covishield, la vacuna que India lanzó con Oxford-AstraZeneca
- El gobierno establece los requisitos para la producción de cannabis medicinal
- Anmat aprobó en el país la primera vacuna que protege contra el dengue
- Ibupirac declarado apto para celíacos
- Comunicación de Novo Nordisk: Diferencias en la indicación de Victoza® y Saxenda®