

Hoy en Revista Dosis
Mostrando artículos por etiqueta: FDA
Lunes, 18 Diciembre 2017 16:03
La FDA aumentará el control sobre los medicamentos homeopáticos

La agencia regulatoria de Estados Unidos publicó hoy un comunicado en el que advierte que “continúa descubriendo que algunos medicamentos homeopáticos se fabrican con ingredientes activos que pueden crear riesgos para la salud sin brindar beneficios médicos comprobados”.
“Hoy, la Administración de Drogas y Alimentos de EE. UU. propuso un nuevo enfoque de aplicación basado en el riesgo para los productos farmacéuticos etiquetados como homeopáticos. Para proteger a los consumidores que optan por utilizar estos productos, el nuevo enfoque actualiza la política de la FDA para abordar mejor las situaciones donde los tratamientos homeopáticos se comercializan para enfermedades y / o condiciones graves, pero donde no se ha demostrado que los productos ofrezcan beneficios clínicos. También cubre situaciones en las que los productos etiquetados como homeopáticos contienen ingredientes potencialmente dañinos o no cumplen con las buenas prácticas de fabricación actuales.
Conforme a la ley, los medicamentos homeopáticos están sujetos a los mismos requisitos relacionados con la aprobación, la adulteración y la rotulación errónea como cualquier otro producto farmacéutico. Sin embargo, los medicamentos recetados y no recetados etiquetados como homeopáticos han sido fabricados y distribuidos sin la aprobación de la FDA bajo las políticas de aplicación de la agencia desde 1988.
“En los últimos años, hemos visto un gran repunte en los productos etiquetados como homeopáticos que se comercializan para una amplia gama de enfermedades y afecciones, desde el resfriado común hasta el cáncer. En muchos casos, las personas pueden depositar su confianza y dinero en terapias que pueden aportar poco o ningún beneficio en la lucha contra dolencias graves o, lo que es peor, pueden causar daños importantes e incluso irreparables porque los productos están fabricados de manera deficiente o contienen ingredientes activos que no están adecuadamente probados”, dijo el comisionado de la FDA, Scott Gottlieb. “Nuestro enfoque para regular los medicamentos homeopáticos debe evolucionar para reflejar la complejidad actual del mercado, al adoptar un enfoque de cumplimiento basado en el riesgo. Respetamos que algunas personas quieran usar tratamientos alternativos, pero la FDA tiene la responsabilidad de proteger al público de los productos que pueden no ofrecer ningún beneficio y tienen el potencial de causar daño”.
Según el nuevo enfoque, es probable que muchos productos homeopáticos queden fuera de las categorías basadas en el riesgo descritas en el nuevo borrador de orientación y que permanezcan disponibles para los consumidores. La FDA tiene la intención de centrar su control en los siguientes tipos de productos:
- Productos con problemas de seguridad reportados;
- Productos que contienen o afirman contener ingredientes asociados con problemas de seguridad potencialmente significativos;
- Productos para vías de administración que no sean orales ni tópicas;
- Productos destinados a ser utilizados para la prevención o el tratamiento de enfermedades y afecciones graves y / o potencialmente mortales;
- Productos para poblaciones vulnerables; y
- Productos que no cumplen con los estándares de calidad, resistencia o pureza requeridos por la ley.
Por ejemplo, los productos que pueden estar sujetos a las nuevas disposiciones son los productos para bebés y niños que contienen ingredientes asociados a problemas de seguridad potencialmente significativos, como belladonna y nux vomica; y productos comercializados para enfermedades graves, como cáncer y enfermedades del corazón.
Dadas las preocupaciones sobre la proliferación de productos potencialmente ineficaces y nocivos etiquetados como homeopáticos, la FDA considerará la adopción de medidas reguladoras y / o coercitivas adicionales, consistentes con las políticas de cumplimiento actuales para proteger al público.
En septiembre de 2016, la FDA advirtió contra el uso de tabletas y geles homeopáticos que contienen belladona, una sustancia tóxica que tiene una respuesta impredecible en niños menores de dos años de edad, después de que los productos se asociaron a eventos adversos graves, incluyendo convulsiones y muertes en bebés y niños. Un análisis de laboratorio de la FDA más tarde confirmó que ciertas tabletas homeopáticas para la dentición contenían niveles elevados e inconsistentes de belladona. Un problema similar ocurrió en 2010 cuando se descubrió que las tabletas para la dentición Hyland’s contenían cantidades variables de belladona. Una inspección de la FDA de las instalaciones de fabricación de ese producto indicó un control deficiente de la fabricación del producto.
La FDA ha emitido advertencias relacionadas con una serie de otros ingredientes de medicamentos homeopáticos en los últimos años. Estos incluyen ciertos productos homeopáticos que contienen zinc que pueden causar pérdida del sentido del olfato, productos para el asma homeopáticos que no han demostrado ser efectivos y varios medicamentos homeopáticos etiquetados para contener ingredientes potencialmente tóxicos, como la nuez vómica, que contiene estricnina (un veneno altamente tóxico y bien estudiado que a menudo se usa para matar roedores).
“Los productos homeopáticos no han sido aprobados por la FDA para ningún uso y pueden no cumplir con los estándares modernos de seguridad, eficacia y calidad”, dijo Janet Woodcock, directora del Centro para la Evaluación e Investigación de Medicamentos de la FDA. “La guía preliminar es un importante paso adelante en el trabajo de la agencia para proteger a los pacientes de productos no probados y potencialmente peligrosos”.
En abril de 2015, la FDA convocó a una audiencia pública para obtener aportes de las partes interesadas sobre el uso actual de productos farmacéuticos etiquetados como homeopáticos, así como el marco regulatorio de la agencia para estos productos. Como resultado de la evaluación de la agencia, que incluyó la consideración de la información obtenida de la audiencia pública y los más de 9.000 comentarios recibidos en el expediente público de la agencia, la FDA ha determinado que es en el mejor interés de la salud pública emitir un nuevo borrador de orientación que propone un enfoque integral de cumplimiento basado en el riesgo para los productos farmacéuticos etiquetados como homeopáticos y comercializados sin la aprobación de la FDA”.
Publicado en
Noticias
Viernes, 15 Diciembre 2017 13:56
FDA autoriza la primera prueba que obtiene el perfil genómico completo de tumores sólidos

|
La Agencia estadounidense del Medicamento (FDA) dio luz verde al uso de la prueba ‘FoundationOne CDx’, desarrollada por la compañía Foundation Medicine y comercializada fuera de Estados Unidos por Roche, que permite obtener el perfil genómico completo de tumores sólidos. |
||
|
|
||
| La aprobación se ha basado en su validación clínica y analítica y, a partir de ahora, podrá usarse como un sistema de diagnóstico complementario para la selección de terapias dirigidas para los pacientes diagnosticados de tumores sólidos.
En concreto, permite evaluar las cuatro tipos de cambios genómicos completos en los 324 genes conocidos como causantes de tumores sólidos, y permite identificar pacientes con cáncer avanzado que probablemente responderán a terapias dirigidas según su perfil genómico individual. Además, permite informar de las características genómicas, incluida la inestabilidad de microsatélites (MSI) y la carga mutacional del tumor (TMB), y las de alteraciones en otros genes (relevantes para otras terapias). De este modo, los médicos puedan utilizar dicha información en el abordaje de cada tumor atendiendo a los criterios de las guías clínicas en oncología. De las 17 terapias actualmente aprobadas para su inclusión en el informe técnico, 12 lo están como tratamiento de primera línea para sus respectivas indicaciones. Por ello, se espera que el número de terapias dirigidas registradas aumente a medida que Foundation Medicine y sus socios obtengan la aprobación de la FDA para incluir biomarcadores adicionales en la plataforma. La farmacéutica suiza Roche, que en 2015 adquirió una participación mayoritaria en Foundation Medicine y ha lanzado la prueba en más de 20 países, ha reconocido que la prueba es el primer sistema de diagnóstico complementario basado en secuenciación de segunda generación para su fármaco alectinib, comercializado como ‘Alecensa’ como tratamiento en monoterapia del cáncer de pulmón no microcítico metastásico ALK+. “Supone un gran avance en la personalización del abordaje oncológico, ya que facilita que los pacientes de Estados Unidos puedan acceder a una prueba diagnóstica complementaria que permite identificar opciones de tratamiento basadas en el perfil molecular del tumor de cada individuo”, ha explicado la responsable de Desarrollo Global de Productos de Roche, Sandra Horning. Fuente: PM Farma |
Publicado en
Noticias
Jueves, 16 Noviembre 2017 14:04
Dispositivo reduce síntomas de abstinencia a opiáceos
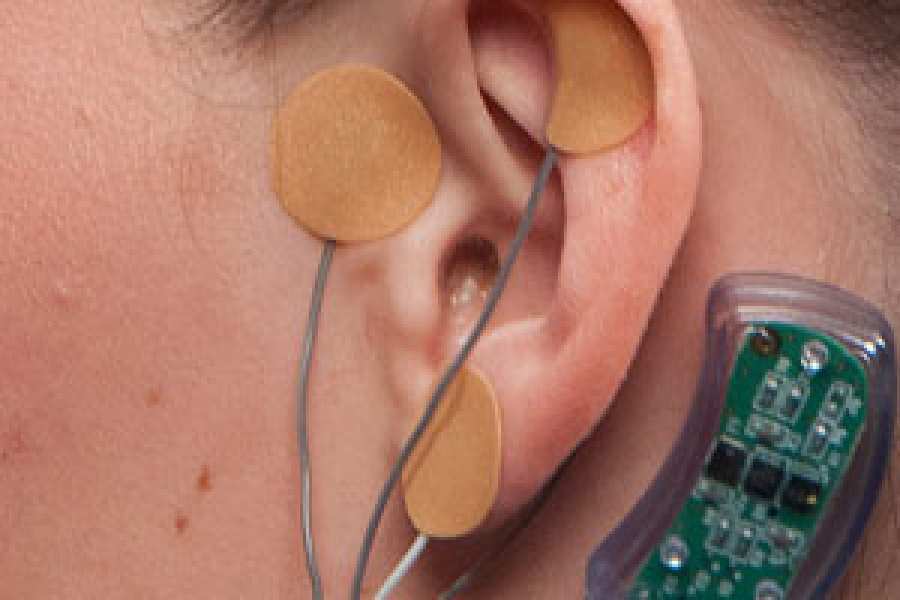
Con la aprobación de su nueva indicación, el dispositivo de estimulación eléctrica NSS-2 Bridge de Innovative Health Solutions se puede utilizar en pacientes hasta por 5 días durante la fase de abstinencia física aguda, para aliviar los síntomas como sudoración, malestar gastrointestinal y agitación, insomnio y dolor en las articulaciones. El dispositivo es un pequeño estimulador eléctrico de nervios que se coloca detrás de la oreja del paciente y emite pulsos eléctricos para estimular las ramas de ciertos nervios craneales.
La aprobación se basa en datos de un estudio clínico de rama única revisada por la FDA. En el estudio, se evaluaron 73 pacientes sometidos a abstinencia física de opioides en función de su escala clínica de abstinencia de opiáceos (COWS). COWS es una evaluación clínica que mide los síntomas de abstinencia de opioides, como la frecuencia de pulso en reposo, sudoración, tamaño de la pupila, problemas gastrointestinales, dolor de huesos y articulaciones, temblores y ansiedad. Las puntuaciones oscilan entre 0 y más de 36, y las puntuaciones más altas representan síntomas más graves.
El puntaje promedio de COWS para los pacientes en el estudio antes de usar el dispositivo fue 20.1. Dentro de los 30 minutos de usar el dispositivo, los pacientes mostraron una reducción en los COWS de al menos 31%. El ochenta y ocho por ciento de los participantes en el ensayo cambiaron a terapia asistida con medicación después de 5 días de usar el dispositivo, junto con los medicamentos necesarios para síntomas persistentes, como náuseas y vómitos.
Este dispositivo ya fue aprobado por la FDA en 2014 para el uso en acupuntura.
Referencias
FDA grants marketing authorization of the first device for use in helping to reduce the symptoms of opioid withdrawal [news release]. FDA’s website. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm585271.htm?utm_campaign=11152017_PR_FDA%20allows%20marketing%20of%20opioid%20withdrawal%20device&utm_medium=email&utm_source=Eloqua.
Publicado en
Noticias
Martes, 07 Noviembre 2017 14:04
Tratamiento aprobado para extraño tipo de cáncer

La FDA extendió la aprobación de vemurafenib (Zelboraf, Genentech) para incluir el tratamiento de ciertos pacientes adultos con Enfermedad de Erdheim-Chester (EEC), un cáncer de sangre poco común, lo que lo convierte en el primer tratamiento aprobado para EEC.
Vemurafenib está indicado para tratar pacientes cuyas células cancerosas tienen una mutación genética específica conocida como BRAF V600. Se aprobó previamente en 2011 para tratar a ciertos pacientes con melanoma que tienen la mutación BRAF V600E. La terapia es un inhibidor de la quinasa que actúa bloqueando ciertas enzimas que promueven el crecimiento celular.
La EEC se origina en la médula ósea y causa un aumento en la producción de histiocitos, un tipo de glóbulo blanco que puede dar lugar a tumores que se infiltran en muchos órganos y tejidos de todo el cuerpo. Actualmente, los pacientes con EEC tienen expectativa de vida muy limitada.
La aprobación se basa en datos del ensayo clínico VE-BASKET de fase 2 que incluyó 22 pacientes con EEC con mutación BRAF-V600 positiva. El ensayo evaluó la eficacia de vemurafenib para el tratamiento de EEC midiendo el porcentaje de pacientes que experimentaron una reducción total o parcial del tamaño del tumor (tasa de respuesta global). El cincuenta por ciento de los pacientes experimentó una respuesta parcial y el 4.5% experimentó una respuesta completa.
Las reacciones adversas graves más comunes informadas en el ensayo incluyeron el desarrollo de nuevos cánceres de piel, presión arterial alta, sarpullido y dolor articular.
Otros efectos adversos comunes incluyen pequeñas protuberancias, pérdida de cabello, cambios en la actividad eléctrica del corazón y crecimientos de la piel.
Referencias
FDA Approves Zelboraf (Vemurafenib) for Erdheim-Chester Disease with BRAF V600 Mutation [news release]. San Francisco. Genentech’s website. https://www.gene.com/media/press-releases/14688/2017-11-06/fda-approves-zelboraf-vemurafenib-for-er. Accessed November 6, 2017
FDA approves first treatment for certain patients with Erdheim-Chester Disease, a rare blood cancer [news release]. FDA’s website. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm583931.htm?utm_campaign=11062017_PR_FDA%20approves%20treatment%20Erdheim-Chester&utm_medium=email&utm_source=Eloqua. Accessed November 6, 2017.
Publicado en
Noticias
Miércoles, 01 Noviembre 2017 14:06
La FDA da un paso hacia las inspecciones globales de fabricación farmacéutica

La FDA reconocerá a 8 autoridades reguladoras europeas de medicamentos, en Austria, Croacia, Francia, Italia, Malta, España, Suecia y el Reino Unido, como capaces de realizar inspecciones de instalaciones de fabricación que cumplan con los requisitos de la FDA, según un comunicado de la agencia publicado hoy.
Este logro marca un hito importante para la implementación y operatividad exitosa del Anexo Farmacéutico modificado del Acuerdo de Reconocimiento Mutuo (MRA) de la Unión Europea (UE) de 1998 que permite a los reguladores de EE. UU. y la UE utilizar las inspecciones de buenas prácticas de fabricación de las instalaciones de fabricación farmacéutica.
Algunas terapias aprobadas en los EE. UU. se fabrican totalmente en el extranjero o se fabrican en los EE.UU. pero contienen algunos ingredientes importados. Todos los medicamentos aprobados en los EE.UU., independientemente de dónde se hayan fabricado, deben cumplir con las normativas vigentes. Una forma en que la FDA supervisa la fabricación de medicamentos es inspeccionando rutinariamente las plantas de fabricación de medicamentos nacionales y extranjeras para verificar el cumplimiento de los estándares de fabricación que garantizan la calidad y los requisitos de la etiqueta del producto.
“A partir del 1 de noviembre, daremos un paso adelante sin precedentes en la concreción de los beneficios clave del Acuerdo de Reconocimiento Mutuo con nuestras contrapartes europeas, ya que ahora confiaremos en los datos de inspección obtenidos por estas 8 agencias reguladoras”, dijo en un comunicado Dara Corrigan, comisionada adjunta en funciones para las operaciones y políticas regulatorias globales de la FDA. “El progreso realizado hasta ahora nos pone en camino para cumplir nuestro objetivo de completar las 28 evaluaciones de capacidades en la UE en julio de 2019″.
En junio de 2017, la Comisión Europea determinó que la FDA “tiene la capacidad y los procedimientos establecidos para llevar a cabo inspecciones GMP a un nivel equivalente a la UE”. La finalización de estas evaluaciones de capacidades permite a la FDA y la UE evitar la duplicación de las inspecciones de drogas y permite a los reguladores dedicar más recursos a la inspección de otras instalaciones de fabricación en los países donde puede haber un mayor riesgo.
Fuente: Pharmacy Times
Publicado en
Noticias
Lunes, 23 Octubre 2017 13:36
La FDA aprueba vacuna contra el Herpes Zoster

La agencia regulatoria de Estados Unidos aprobó la comercialización de la vacuna zóster recombinante, adyuvada (Shingrix, GlaxoSmithKline) para la prevención del herpes zoster en adultos de 50 años o más. Shingrix es una vacuna de subunidad recombinante inactivada, administrada por vía intramuscular en dos dosis.1
La aprobación se basó en ensayos clínicos de fase 3 que evaluaron la eficacia, seguridad e inmunogenicidad de la vacuna en más de 38.000 personas. En un análisis conjunto de estos estudios, Shingrix demostró eficacia mayor al 90% contra el herpes zóster en todos los grupos de edad, así como una eficacia sostenida durante un período de seguimiento de 4 años2,3. Al prevenir el herpes zóster, Shingrix también redujo la incidencia global de la neuralgia posherpética (NPH), una forma de dolor crónico del nervio y la complicación más común asociada con el herpes zóster.
Se espera que el Comité Asesor sobre Prácticas de Inmunización (ACIP) del Centro para el Control y la Prevención de Enfermedades de EE. UU. votenuna recomendación para el uso de Shingrix en su reunión a fines de esta semana.
Tras esta aprobación de la FDA, y en espera de una recomendación de ACIP, Shingrix estará disponible en Estados Unidos.
Referencias
1 Shingrix approved in the US for prevention of shingles in adults aged 50 and over [news release]. London. GlaxoSmithKline press office. October 20, 2017. https://www.gsk.com/en-gb/media/press-releases/shingrix-approved-in-the-us-for-prevention-of-shingles-in-adults-aged-50-and-over/. Accessed Oct. 21, 2017.
2 Lal H et al. Efficacy of an Adjuvanted Herpes Zoster Subunit Vaccine in Older Adults. N Engl J Med.2015;372:2087-96.
3 Cunningham et al. Efficacy of the herpes zoster subunit vaccine in adults 70 years of age or older. N Engl J Med. 2016;375:1019-32
Fuente: Pharmacy Times
Publicado en
Noticias
Jueves, 19 Octubre 2017 14:32
La FDA aprueba la segunda terapia de células CAR-T

Axicabtagene ciloleucel (Yescarta, Kite Phama) recibió la aprobación de la agencia regulatoria estadounidense para el tratamiento de pacientes con linfoma no Hodgkin (LNH) de células B agresivo recidivante / refractario que no son elegibles para el trasplante autólogo de células madre.
Esta aprobación se da después de la de tisagenlecleucel-T (Kymriah, Novartis), la primera terapia en ingresar al mercado que usa la tecnología CAR. Indicado para uso en pacientes pediátricos y adultos jóvenes (de 3 a 25 años) con leucemia linfoblástica aguda recidivante o refractaria (LLA), el producto recibió la luz verde el 30 de agosto.
La terapia implica la ingeniería de las células T de un paciente para expresar un CAR que luego direccionará al antígeno CD19, una proteína expresada en la superficie celular de linfomas de células B y leucemias. Luego, las células son redirigidas para matar las células cancerosas.
Las células CAR T han demostrado resultados en ensayos clínicos en pacientes con cánceres hematológicos recidivantes / refractarios, que tienen opciones terapéuticas limitadas o carecen de ellas. En algunos casos, una dosis del tratamiento ha erradicado la enfermedad.
Alta tasa de respuesta
La aprobación de hoy se basa en los datos del ensayo pivotal ZUMA-1 para axicabtagene ciloleucel, que se realizó en 101 pacientes con LNH agresivo, en colaboración con el Instituto Nacional del Cáncer. La compañía publicó los resultados finales en febrero de 2017, http://ir.kitepharma.com/releasedetail.cfm?ReleaseID=1014817, que mostró una tasa de respuesta general (ORR) en el 82% de los pacientes y una respuesta completa (CR) en el 54% .
A los 6 meses, el ORR se mantuvo en el 41% de los pacientes y CR en el 36%.
Además, cinco de los 101 pacientes (5%) continúan experimentando respuestas parciales (PR) altamente significativas y duraderas con mínimas anomalías en las tomografías por emisión de positrones. Uno de estos RP se convirtió a un CR en el mes 9, informó la compañía.
El fabricante aún no ha anunciado cuánto costará este nuevo tratamiento. Las inmunoterapias no solo han abierto nuevos caminos en innovación sino también en costos. Novartis había anunciado que tisagenlecleucel-T tendría un precio de $ 475.000, lo que bien podría estar probando los límites de lo que las compañías de seguros y los pacientes pueden estar dispuestos a pagar, pero también se ha señalado que esta es una terapia “única potencialmente curativa”. En casos exitosos, el tratamiento de una sola vez puede erradicar la enfermedad. El paralelo más cercano son los trasplantes de médula ósea, que cuestan alrededor de $ 800.000 para un trasplante alogénico de médula ósea en los Estados Unidos y alrededor de $ 350.000 para un trasplante autólogo, aunque estos costos varían considerablemente.
Riesgo de eventos adversos graves
Sin embargo, estas terapias pueden asociarse con una toxicidad grave, y se han producido muertes relacionadas con el tratamiento en ensayos clínicos. Este riesgo había llevado a restringir el uso de la terapia a centros especialmente certificados.
En ensayos clínicos, CRS se ha tratado con éxito con un bloqueador de interleucina-6, tocilizumab (Actemra, Genentech), que ya se comercializa para su uso en la artritis reumatoide. La FDA ahora ha ampliado su indicación para incluir el tratamiento de CRS grave o potencialmente mortal inducido por células T CAR en pacientes de 2 años de edad o mayores. La agencia notó que en los ensayos clínicos entre pacientes tratados con células T CAR, el 69% de los pacientes tenían resolución completa de CRS dentro de 2 semanas después de una o dos dosis de tocilizumab.
El riesgo de estas toxicidades agudas únicas, que son infrecuentes con otras terapias contra el cáncer, ha impulsado el desarrollo de un marco para tratarlas sistemáticamente. Los médicos del Centro Oncológico MD Anderson de la Universidad de Texas en Houston elaboraron una guía que aborda el síndrome de encefalopatía relacionada con las células T y CRS (CRES).
También se han observado otros efectos secundarios graves con la terapia con células T CAR. En el ensayo ZUMA-1, se incluyeron anemia (43%), neutropenia (39%), disminución del recuento de neutrófilos (32%), neutropenia febril (31%), recuento de glóbulos blancos (29%), trombocitopenia (24% ), encefalopatía (21%) y disminución del recuento de linfocitos (20%). Se observó CRS de grado 3 o superior en el 13% de los pacientes y eventos neurológicos en el 28%, pero no hubo casos de edema cerebral.
Debido al riesgo de eventos adversos graves, axicabtagene ciloleucel ha sido aprobado con una estrategia de evaluación y mitigación de riesgos (REMS), que incluye elementos para garantizar un uso seguro (ETASU). La FDA ha restringido el uso de estos agentes a centros especialmente certificados. La certificación requeriría que todo el personal involucrado en la prescripción, dispensación o administración de la terapia de células CAR T se capacite para reconocer y gestionar los CRS y los eventos neurológicos. Además, los pacientes también deben estar informados de los posibles efectos secundarios graves y de la importancia de regresar rápidamente al sitio de tratamiento si se desarrollan efectos secundarios.
Para evaluar más a fondo la seguridad a largo plazo, la FDA también exige que el fabricante realice un estudio observacional posterior a la comercialización.
Publicado en
Noticias
Martes, 10 Octubre 2017 13:56
FDA aprueba device para apnea

Se trata de Remedē System, un dispositivo implantable que monitorea las señales respiratorias del paciente durante el sueño y estimula un nervio para mover el diafragma y restablecer la respiración normal.
El producto, que recibió el OK del organismo para ser utilizado en personas con apnea central del sueño, de moderada a severa, es implantado por un cardiólogo sin el uso de anestesia general.
Remedē System lleva la firma de la norteamericana Respicardia, una compañía enfocada en terapias cardiovasculares y respiratorias.
Fuente: Pharmabiz
Publicado en
Noticias
Martes, 22 Agosto 2017 03:00
Aprobación

La Administración de Drogas y Alimentos de los Estados Unidos (FDA, por sus siglas en inglés) aprobó Duzallo (Ironwood Pharmaceuticals), una combinación oral de dosis fija de lesinurad (Zurampic, AstraZeneca) y alopurinol (varias marcas) para el tratamiento de la hiperuricemia asociada con la gota en pacientes cuyos niveles séricos de ácido úrico (sUA) no se han logrado alcanzar con el alopurinol solo.
Duzallo en dosis diaria contiene lesinurad 200 mg más alopurinol 300 mg. También estará disponible en una dosis de 200 mg de lesinurad más 200 mg de alopurinol.
Este medicamento no se recomienda para el tratamiento de la hiperuricemia asintomática.
El alopurinol reduce la producción de ácido úrico, y lesinurad aumenta la excreción renal de AU. La FDA aprobó lesinurad en 2015.
En ensayos clínicos de adultos con gota, para los cuales los niveles objetivo de sUA no pudieron alcanzarse con el alopurinol solo, lesinurad en combinación con alopurinol casi duplicó el número de pacientes en los que se alcanzó el objetivo de sUA <6 mg / dL a los 6 meses. Redujo el nivel medio de sUA a <6 mg / dL por 1 mes y mantuvo ese nivel a través de 12 meses, informó la compañía que desarrolló el producto.
Las reacciones adversas más comunes en los ensayos clínicos fueron dolor de cabeza, gripe, aumento de los niveles de creatinina sanguínea y reflujo ácido. Duzallo tiene una advertencia destacada con respecto al riesgo de insuficiencia renal aguda.
El medicamento debe tomarse por la mañana con alimentos y agua, y se debe aconsejar a los pacientes que se mantengan bien hidratados, y que tomen 2 litros de líquido al día al seguir el tratamiento.
Publicado en
Noticias
Martes, 08 Agosto 2017 02:19
La FDA aprueba un tratamiento mensual inyectable para el trastorno bipolar

La formulación de liberación prolongada de aripiprazol (Abilify Maintena, Otsuka Pharmaceutical Co, Ltd y H. Lundbeck A / S) fue aprobada por la Food and Drug Administration de los Estados Unidos (FDA) para su uso como suspensión inyectable para monoterapia de mantenimiento de desorden bipolar I (BP-I) en adultos, anunciaron las compañías.
Aripiprazol, un antipsicótico atípico, es un polvo liofilizado estéril que, cuando se reconstituye con agua estéril, forma una suspensión que se puede administrar por inyección. Fue creado por Otsuka en Japón y ha sido desarrollado y comercializado conjuntamente por Otsuka y Lundbeck.
“Es una nueva opción de tratamiento para los pacientes que han establecido tolerabilidad con aripiprazol oral”, afirma el Dr. Joseph Calabrese, MD, director del Programa de trastornos del estado de ánimo en el Centro Médico de la Universidad de Cleveland en un comunicado de prensa.
“Recibir aripiprazol cada mes según lo prescrito y administrado por un profesional de la salud proporciona a los pacientes la oportunidad de estar libres de tomar su antipsicótico diariamente para tratar el trastorno bipolar I”.
El especialista hizo hincapié en que la medicación antipsicótica oral concomitante debe administrarse durante 14 días después de la primera inyección.
Enfermedad crónica
BP-I es una enfermedad mental crónica. La prevalencia de 12 meses es del 1,5%, y la prevalencia a lo largo de la vida es del 2,1%, según la información proporcionada por la empresa. Los pacientes con BP-I experimentan uno o más episodios de manía y pueden tener episodios de manía y depresión.
El ensayo fase 3, doble ciego, aleatorizado controlado con placebo que apoyó la aprobación reglamentaria demostró la eficacia y la seguridad de Abilify Maintena en la monoterapia de mantenimiento de BP-I. El estudio incluyó pacientes de 18 a 65 años que estaban experimentando un episodio maníaco y que cumplieron con los criterios de DSM-IV-TR para el trastorno bipolar I.
Los pacientes tenían antecedentes de al menos un episodio maníaco o mixto anterior con síntomas maníacos suficientemente graves como para requerir hospitalización y/o tratamiento con un estabilizador del estado de ánimo y/o tratamiento con un agente antipsicótico.
Las condiciones de los participantes del estudio se habían estabilizado con Abilify Maintena antes de la aleatorización.
El estudio encontró que el fármaco retrasó significativamente el tiempo hasta la recurrencia de cualquier episodio de estado de ánimo durante un período de tratamiento de 52 semanas en comparación con placebo. Hubo diferencias significativas entre los grupos de tratamiento al retrasar el tiempo hasta la recurrencia de los episodios maníaco y mixto, pero no hubo diferencias sustanciales en los episodios de humor depresivo.
El mecanismo de la eficacia de este fármaco es desconocido, pero se informó que el fármaco puede actuar a través de una combinación de actividad agonista parcial en la dopamina D2 y receptores 5-HT1A de la serotonina y actividad antagonista en los receptores 5-HT2A.
Eventos adversos
Abilify Maintena fue aprobado en los Estados Unidos en 2013 para el tratamiento de adultos con esquizofrenia. Las reacciones adversas más comunes con el fármaco en pacientes con esquizofrenia son aumento de peso, acatisia, dolor en el lugar de la inyección y sedación.
Según la información de antecedentes en el comunicado de prensa de la compañía, los pacientes ancianos con psicosis relacionada con demencia que se tratan con fármacos antipsicóticos tienen un mayor riesgo de muerte. Abilify Maintena no está aprobado para el tratamiento de pacientes con psicosis relacionada con demencia. El fármaco está contraindicado en pacientes con hipersensibilidad conocida al aripiprazol.
El comunicado enumera otros posibles eventos adversos y destaca un aumento en la incidencia de eventos adversos cerebrovasculares (por ejemplo, accidente cerebrovascular y ataque isquémico transitorio) en pacientes ancianos con psicosis relacionada con demencia que se someten a tratamiento con aripiprazol oral.
El síndrome neuroléptico maligno es un complejo de síntomas potencialmente fatal reportado en asociación con el uso de Abilify Maintena y otros fármacos antipsicóticos. Se cree que el riesgo de disquinesia tardía aumenta con la duración del tratamiento y la dosis acumulativa total de fármacos antipsicóticos, señalaron las empresas.
Los fármacos antipsicóticos atípicos también se han relacionado con cambios metabólicos, incluyendo hiperglucemia y diabetes. En pacientes que toman aripiprazol se han notificado impulsos intensos, particularmente en el juego, y la incapacidad para controlar estos impulsos.
También se advirtió que Abilify Maintena puede causar hipotensión ortostática y debe utilizarse con precaución en pacientes con enfermedad cardiovascular conocida, enfermedad cerebrovascular o condiciones que los predispondrían a la hipotensión.
Fuente: Medscape
Publicado en
Noticias


Lo más visto
- Covishield, la vacuna que India lanzó con Oxford-AstraZeneca
- El gobierno establece los requisitos para la producción de cannabis medicinal
- Anmat aprobó en el país la primera vacuna que protege contra el dengue
- Ibupirac declarado apto para celíacos
- Comunicación de Novo Nordisk: Diferencias en la indicación de Victoza® y Saxenda®