

Hoy en Revista Dosis
Mostrando artículos por etiqueta: Europa
Viernes, 28 Enero 2022 13:30
La píldora de Pfizer contra el Covid-19 fue aprobada en Europa

La Agencia Europea de Medicamentos (EMA) aprobó la píldora anticovid del laboratorio Pfizer, que se convierte en el primer tratamiento oral contra el covid-19 autorizado en la Unión Europea (UE).
“El Comité de Medicamentos de Uso Humano (CHMP) de la EMA ha recomendado la concesión de una autorización condicional de comercialización para el medicamento antiviral oral Paxlovid”, declaró el regulador europeo en un comunicado.
La EMA “recomendó autorizar el Paxlovid para el tratamiento del covid-19 en los adultos que no necesitan asistencia respiratoria pero que presentan un riesgo de que la enfermedad se agrave”.
Los antivirales actúan reduciendo la capacidad de un virus para replicarse, frenando así la enfermedad.
Son muy esperados porque son fáciles de administrar, pudiendo ser simplemente ingeridos con un vaso de agua.
Pfizer había declarado en diciembre que su píldora anticovid reducía en casi 90% las hospitalizaciones y muertes en personas de riesgo cuando era ingerida en los primeros días después de la aparición de los síntomas.
La EMA evaluó los datos de un estudio en el que participaron pacientes con covid-19 que demostraron que “el tratamiento con Paxlovid redujo significativamente las hospitalizaciones o muertes en pacientes que presentaban al menos una enfermedad subyacente que los exponía a un riesgo de contraer un covid-19 grave”.
La mayoría de los pacientes del estudio estaban infectados con la variante Delta, indicó la Agencia Europea de Medicamentos, pero señaló que según las pruebas de laboratorio, Paxlovid debería seguir siendo eficaz contra Ómicron.
La píldora Pfizer es una combinación de una nueva molécula, PF-07321332, y ritonavir, un antiviral contra el VIH, que se toman en forma de comprimidos separados.
“El Comité de Medicamentos de Uso Humano de la EMA concluyó que los beneficios del medicamento son mayores que sus riesgos para el uso aprobado y enviará sus recomendaciones a la Comisión Europea (CE) para una decisión rápida, aplicable en todos los Estados miembros de la UE”.
Este procedimiento de aprobación por parte de la CE suele llevar algunas horas o días.
“Con la autorización de Paxlovid esta semana, se han autorizado 6 medicamentos anticoronavirus en el marco de la estrategia terapéutica de la UE”, dijo en otro comunicado la comisaria europea de Salud y Seguridad Alimentaria, Stella Kyriakides.
Estados Unidos, Canadá e Israel forman parte de un puñado de países que ya han dado luz verde al tratamiento de Pfizer.
Publicado en
Noticias
Jueves, 19 Septiembre 2019 15:25
Europa: 50% de ensayos para la aprobación de oncológicos plantea dudas de sesgo

Según una nueva investigación publicada por The BMJ, alrededor de la mitad de los ensayos clínicos que respaldaron las nuevas aprobaciones de medicamentos contra el cáncer en Europa entre 2014 y 2016 tenían un alto riesgo de sesgo, lo que indica que los efectos del tratamiento podrían haber sido exagerados.
Los hallazgos agregan peso a la investigación existente que plantea serias preocupaciones sobre los bajos estándares de evidencia que respaldan los nuevos medicamentos contra el cáncer, y resaltan la necesidad de mejorar el diseño, la realización, el análisis y la notificación de los ensayos de medicamentos contra el cáncer.
Fallos en el diseño
En 2017, más de una cuarta parte (24 de 92) de las aprobaciones de la Agencia Europea del Medicamento (EMA) fueron para fármacos contra el cáncer, la mayoría de los cuales se basaron en pruebas de ensayos controlados aleatorios, considerados el estándar de oro para evaluar la efectividad del tratamiento.
Sin embargo, los fallos en el diseño, el desarrollo, el análisis o el informe de los ensayos controlados aleatorios pueden distorsionar las estimaciones del efecto del tratamiento, lo que puede poner en peligro la validez de sus hallazgos.
Riesgo de sesgo
Para evaluar estos defectos con más detalle, un equipo de investigadores internacionales liderado por Huseyin Naci, profesor asistente de Política de Salud en el Departamento de Política de Salud del London School of Economics and Political Science, en Londres, examinó el diseño, el riesgo de sesgo y la notificación de ensayos controlados aleatorios que respaldaron las aprobaciones europeas de medicamentos contra el cáncer entre 2014 y 2016.
Los reguladores identificaron problemas adicionales con 10 de los 32 nuevos medicamentos
Durante este período, la EMA aprobó 32 nuevos medicamentos contra el cáncer sobre la base de 54 estudios. De estos, 41 (76%) fueron ensayos controlados aleatorios; 39 tenían publicaciones disponibles y, por lo tanto, se incluyeron en el estudio.
Solo 10 ensayos (26%) midieron la supervivencia general como criterio de valoración principal (objetivo primario). Los 29 ensayos restantes (74%) evaluaron las medidas indirectas (sustitutivas) del beneficio clínico, que no siempre predicen de manera confiable si un paciente vivirá más o tendrá una mejor calidad de vida.
En general, se consideró que 19 ensayos (49%) tenían un alto riesgo de sesgo debido a déficits en su diseño, conducta o análisis. Los ensayos que evaluaron la supervivencia general tenían menor riesgo de sesgo que los que evaluaron las medidas sustitutivas del beneficio clínico.
Los reguladores identificaron problemas adicionales con 10 de los 32 nuevos medicamentos (31%) aprobados durante este período, pero estas preocupaciones rara vez surgieron en la literatura científica.
Complejidad de los ensayos clínicos
Los investigadores señalan varias limitaciones en la investigación. Por ejemplo, no incluyeron informes de estudios clínicos, que contienen información detallada sobre los métodos y resultados de los ensayos, y se centraron solo en ensayos de medicamentos contra el cáncer, por lo que los resultados pueden no aplicarse a los ensayos en otras áreas terapéuticas.
Además, evaluaron el riesgo de sesgo en lugar del sesgo en sí mismo: sigue siendo una posibilidad que los déficits metodológicos identificados por los autores no conduzcan a resultados sesgados.
También reconocen que algunos de los sesgos pueden ser inevitables debido a la complejidad de los ensayos de cáncer, pero dicen que sus hallazgos deberían incitar a los encargados de formular políticas, los investigadores y los médicos “a considerar cuidadosamente el riesgo de sesgo en los ensayos fundamentales que respaldan las decisiones reguladoras, y el alcance de qué nuevas terapias contra el cáncer ofrecen un beneficio significativo para los pacientes”.
La evidencia inexacta conduce a daños intangibles, lo que fomenta la falsa esperanza y crea una distracción de los cuidados paliativos
Las publicaciones científicas y los documentos reglamentarios deberían facilitar que los pacientes y los médicos comprendan qué tan bien se realiza un estudio”, agregan.
Daños directos e indirectos
En un editorial vinculado al estudio, investigadores con sede en Australia argumentan que la incertidumbre y la exageración de la evidencia que respalda la aprobación de los medicamentos contra el cáncer “causa daño directo si los pacientes corren el riesgo de efectos adversos graves o fatales sin un beneficio probable, o si renuncian a tratamientos más efectivos y seguros”.
La evidencia inexacta también conduce a daños intangibles, lo que fomenta la falsa esperanza y crea una distracción de los cuidados paliativos necesarios, agregan.
Este estudio muestra que la evidencia del ensayo por sí sola no es suficiente, escriben. La evaluación de la calidad de esa evidencia también es necesaria para garantizar que estos ensayos estimen con precisión los efectos del tratamiento.
Fuente: Redacción Médica – España / COFA
Publicado en
Noticias
Miércoles, 13 Febrero 2019 13:46
Europa sienta las bases del proyecto de prospecto farmacológico común

Los altos directos de las Agencias de Medicamentos (HMA), la Agencia Europea del Medicamento (EMA) y la Comisión Europea (CE) lanzaron este martes a consulta pública la creación de una base de datos digital para la puesta en común de los prospectos de los medicamentos.
El proyecto consiste en poner a información del usuario y del profesional, desde una plataforma web, la información de un medicamento de la Unión Europea en el que se incluirá el prospecto para pacientes y el resumen de las características del producto (SPC) para profesionales de la salud.
De esta forma, el prospecto (que actualmente se encuentra en la caja del medicamento y como documento pdf en los sitios web de los reguladores) pasará a formar parte de esta plataforma digital que se ha nombrado bajo las siglas ePI.
Qué es el ePI
Según explica el desglose del proyecto, la ePI aunará la información legal del producto para medicamentos (es decir, SPC, PL y etiquetado) en una formato “organizado” y que permite su difusión a través de Internet, plataformas electrónicas e impresos.
Este nuevo formato “semiestructurado adecuado para manejo electrónico” contendrá algunos elementos estructurados (por ejemplo, fijos títulos y vocabularios), y algunos elementos no estructurados (es decir, texto libre).
El plazo de aportaciones finaliza en julio
“Esto puede abordar algunas de las limitaciones actuales y satisfacer mejor las necesidades de los pacientes y profesionales de la salud para obtener información accesible y actualizada sobre medicamentos en el momento de necesidad”, argumenta la Unión Europea el en documento que han hecho público.
El borrador de principios clave se deriva de acuerdos a cabo por la Agencia Europea del Medicamento (EMA), la HMA y la CE a lo largo de 2018. El periodo de consulta, al que se pueden enviar aportaciones, se cerrará en el mes de julio. Tras dicha consulta, se acordará la versión final de los principios clave. Luego formarán la base para respaldar un enfoque armonizado del ePI en toda la Unión Europea.
Según estos expertos, es muy para la salud pública poner en común la información electrónica de los medicamentos como un complemento al prospecto en papel. De esta forma, se pondrán en común las ePI en todas las lenguas oficiales de la UE.
Fuente: Redacción Médica – España / COFA
Publicado en
Noticias
Martes, 29 Mayo 2018 15:02
Así es la red público-privada que está desarrollando medicamentos pediátricos en Europa

La industria farmacéutica participa en la iniciativa ‘conect4children’ (c4c), una red público-privada para impulsar el desarrollo de medicamentos pediátricos en Europa, en la que participan 33 centros académicos y de investigación y 10 compañías farmacéuticas, y que tendrá una duración de seis años y un presupuesto de 140 millones de euros.
Más de 50 entidades de 20 países europeos se han unido en el consorcio ‘conect4children’.
El coordinador del proyecto, el profesor Carlo Giaquinto, de la Fondazione PENTA Onlus de la Universidad de Pádova (Italia), cree que ‘conect4children’ debe afrontar “serios problemas en cuanto al diseño, desarrollo y realización de ensayos clínicos pediátricos, tales como los esfuerzos fragmentados y duplicados de promotores, entidades y países; la escasez de pacientes disponibles para los ensayos en muchas patologías pediátricas, y la carencia de centros y expertos capacitados para llevar los ensayos a buen puerto
La iniciativa aspira a generar las infraestructuras necesarias que optimicen la realización de ensayos clínicos pediátricos mediante avances como la creación de un único centro de gestión para todos los promotores, centros e investigadores; la gestión eficiente de los ensayos clínicos mediante la adopción de indicadores de calidad homogéneos, enfoques compartidos y la coordinación de centros tanto a nivel nacional como internacional; y la colaboración entre redes nacionales y especializadas.
Además, de la mejora en el diseño y la preparación de los ensayos mediante un estudio de viabilidad estratégico y operacional; la promoción del diseño de ensayos innovadores y la aplicación de métodos científicos cuantitativos; la creación de una plataforma educativa para formar a futuros expertos en el desarrollo de nuevos medicamentos pediátricos, y el desarrollo del soporte necesario para todas estas actividades.
Fuente: Periodista digital
Publicado en
Noticias
Viernes, 02 Marzo 2018 15:44
Llega a Europa un medicamento para tratar tumores ginecológicos
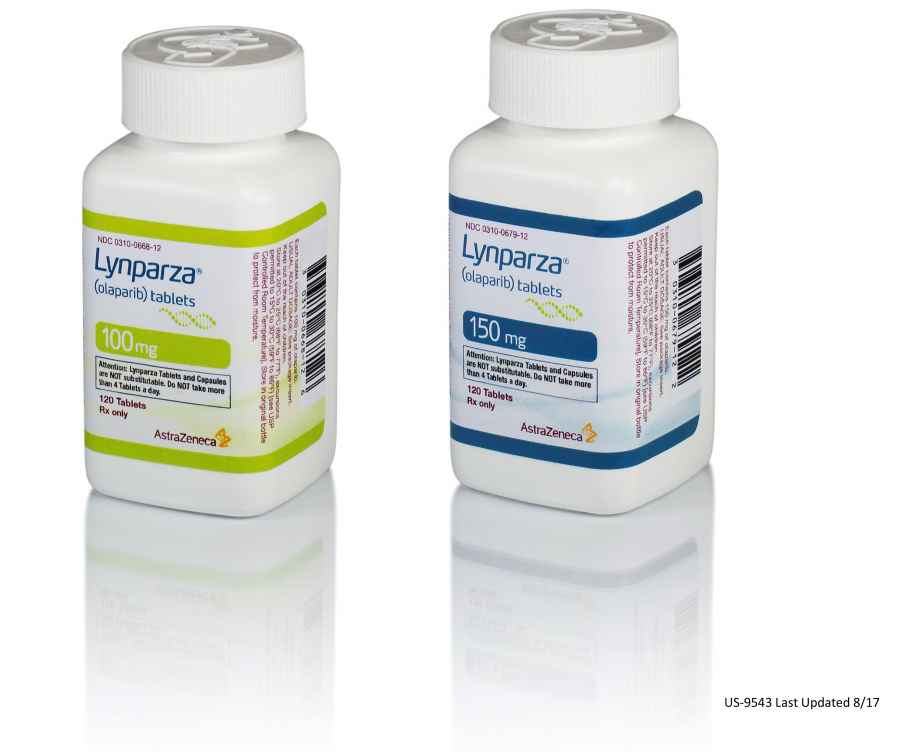
Esta semana, la Agencia Europea de Medicamentos (EMA, por sus siglas en inglés) autorizó el segundo medicamento oncológico de la farmacéutica AstraZeneca para el mercado europeo. Se trata de olaparib, registrado con el nombre comercial de Lynparza, en comprimidos de 300 miligramos, para el tratamiento de mantenimiento de las pacientes con cáncer de ovario epitelial, cáncer de las trompas de falopio o cáncer peritoneal primario en recaída sensible a platino, que presentan respuesta completa o parcial a la quimioterapia basada en platino. Según el informe del Comité de Medicamentos de Uso Humano (CHMP) de esa entidad, el dictamen favorable permite recomendar el producto para estos males ginecológicos.
"Los datos muestran que olaparib consigue un control a largo plazo de la enfermedad, retrasando la necesidad de quimioterapia en este amplio grupo de mujeres con cáncer de ovario en recaída sensible a platino, independientemente de su estado mutacional de BRCA. También ofrece un perfil de seguridad y tolerabilidad bien conocido que es fundamental para que las pacientes continúen el tratamiento", informó el vicepresidente ejecutivo de Desarrollo Global de Medicamentos y director médico de AstraZeneca, Sean Bohen. Se trata del primer inhibidor de la poli ADP-ribosa polimerasa (PARP) aprobado, inicialmente fue autorizado como formulación en cápsula. La nueva formulación en comprimidos permitirá reducir la administración de 8 cápsulas dos veces al día a 2 comprimidos dos veces al día.
La recomendación del CHMP se basa en dos ensayos aleatorizados, SOLO-2 y Estudio 19, que demostraron que olaparib redujo el riesgo de progresión de la enfermedad o muerte en las pacientes en recaída platino-sensibles frente a placebo. "Estamos muy contentos con este dictamen favorable basado en datos que confirman el potencial de olaparib como tratamiento de mantenimiento para las mujeres con cáncer de ovario en recaída sensible a platino. Esperamos continuar nuestra colaboración con AstraZeneca para poner olaparib a disposición de los pacientes en Europa", ha dicho el vicepresidente senior, director de Desarrollo Clínico Global y director médico de Merck, Roy Baynes, socio de la firma en el desarrollo del tratamiento.
Olaparib se comercializa en casi 60 países y ha tratado a más de 20 mil pacientes en todo el mundo. En enero de 2018, fue aprobado en Estados Unidos por la Agencia Americana del Medicamento (FDA, por sus siglas en inglés) para su uso en el cáncer de mama metastásico, convirtiéndose así en el primer inhibidor de PARP autorizado en indicaciones distintas del cáncer de ovario.
*Mirada profesional
Publicado en
Noticias
Martes, 06 Febrero 2018 15:21
Europa: el 30% de los nuevos fármacos aprobados en 2017 son oncológicos
En el marco del informe anual de la Agencia europea de Medicamentos (EMA, según sus siglas en inglés), se informó que de los nuevos tratamientos aprobados en el viejo continente el año pasado el 26 por ciento del conjunto de las 92 opiniones positivas correspondieron a tratamientos contra el cáncer, porcentaje que asciende hasta el 31 por ciento en el caso de los 35 fármacos que incluyen nuevas moléculas. En total, 24 antitumorales recibieron la opinión positiva, de los que 11 son tratamientos que incluyen un principio activo completamente nuevo. Estas opiniones positivas de la EMA se convierten, en un trámite posterior, en decisiones de autorización por parte de la Comisión Europea. Así, en el caso de los 35 fármacos que incluyen nuevas moléculas, un total de 30 ya han sido autorizados, y de ellos el 33% son tratamientos oncológicos.
Los nuevos antitumorales aprobados en Europa se destinan a diversos tipos de tumores, como los cánceres de pulmón, riñón, mama y próstata, el carcinoma de células de Merkel, la leucemia linfoblástica aguda, los tumores neuroendocrinos y la leucemia mieloide aguda, entre otros.
Los datos europeos son coincidentes, en general, con los norteamericanos. Así, el 28 por ciento de los nuevos fármacos aprobados por la FDA estadounidense son tratamientos contra diferentes tipos de cáncer, hasta el punto de que la propia agencia reguladora ha considerado, en su informe anual, que 2017 ha sido "otro año fuerte para la puesta a disposición de los pacientes de nuevos antitumorales". La agencia destaca, además, que 2017 ha sido el año en el que se ha aprobado la primera terapia génica contra un tipo de cáncer, en este caso una forma de leucemia linfoblástica aguda.
Publicado en
Noticias


Lo más visto
- Covishield, la vacuna que India lanzó con Oxford-AstraZeneca
- El gobierno establece los requisitos para la producción de cannabis medicinal
- Anmat aprobó en el país la primera vacuna que protege contra el dengue
- Ibupirac declarado apto para celíacos
- Comunicación de Novo Nordisk: Diferencias en la indicación de Victoza® y Saxenda®